The next decade of biotech innovation will be led by organisations that can produce novel molecules at an economically viable cost.
The biotech industry has crossed a critical threshold. Across AI-driven drug discovery, synthetic biology, diagnostics, and DNA data storage applications, product development success now depends on the capability to rapidly iterate through massive libraries of complex DNA, RNA, and proteins.
However, the capacity to create, test and optimise millions of molecular variants has become the limiting factor for many organisations trying to bring innovative products to market.
Molecular synthesis has evolved into a unique bottleneck for teams at the frontier of biotech R&D. The defining challenge is no longer designing the optimal sequence — it is producing it at the scale, speed, and cost required to turn theoretical potential into a commercially viable reality.
Conventional synthesis platforms were never engineered for this magnitude of throughput. When organisations attempt to scale traditional technologies from thousands to millions of variants, the linear increase in reagent consumption, infrastructure requirements, and operational overhead quickly becomes prohibitive. Overcoming this barrier requires a fundamental departure from legacy processes.
In this white paper, we explore why reaction miniaturisation has emerged as the definitive architecture for high-throughput, low-volume synthesis, and how leading organisations are transforming cost and scalability challenges into competitive advantages, powering the next wave of biotechnology innovation.
Enabling scale-out in biotechnology
Commercial success of a biotechnological process often requires reaction scale-up or scale-out. While scale-up reduces manufacturing costs for products such as antibodies or DNA, scale-out is required to synthesise sets of slightly different products, such as DNA strands, in parallel processes. In the past decade, innovation opportunities in reaction scale-out have emerged across the following areas:
- DNA/RNA oligo and peptide synthesis for drug discovery
- Microarrays for diagnostics and health screening
- DNA data storage
- Synthetic biology
- Enzyme engineering
- Capture/detection probes for targeted sequencing applications.
The common underlying principle of the above applications is the cyclic addition of building blocks (nucleotides, short nucleotide oligos, and amino acids) to a linear growing polymer (DNA/RNA or peptide strand, respectively). An example illustration: high-throughput screening applications require having 105-1012 of DNA/RNA oligonucleotide variant libraries, which are synthesised by repeating a building block addition–wash–deprotection cycle 10s-100s of times.
Scalability of currently available automated DNA oligo synthesis approaches is in the range of 103-105 oligos on column-based DNA oligo synthesisers [1] or robotic well plate liquid handlers [2], which is way below that of some particular current biomolecular synthesis market needs. Different segments of this market have distinct, compounding requirements. In personalised medicine, speed is as critical as scale: a custom capture probe library designed around an individual patient’s tumour must be available within days of diagnosis, because clinical monitoring windows do not accommodate synthesis timelines measured in months. For screening-intensive applications in drug discovery and directed evolution, the constraint is complexity at competitive cost, with libraries spanning 10⁵ to 10¹² variants requiring per-variant economics that column-based or well-plate approaches cannot approach. For write-once archival applications, commercial viability depends on driving cost per synthesised base to levels reachable only through radical miniaturisation. In each application, the ceiling is identical: synthesis economics sets the boundary of what is scientifically reachable and commercially viable within a given budget and timeline.
In a wider context, the DNA, RNA, and protein synthesis markets also face a directional pressure: design-make-test cycles are accelerating, driven in part by AI-assisted platforms that compress the time between hypothesis and experiment. Synthesis throughput is now a rate-limiting step in the innovation cycle. Consequently, each physical iteration takes longer and covers fewer variants than modern computational tools can generate. The resulting deficit in experimental data starves computational models of the feedback necessary for rapid refinement. Resolving this data starvation — particularly in markets such as high-throughput screening and DNA data storage — now depends on testing full, complex libraries using highly scalable automated platforms capable of pushing synthesis scales beyond 10⁵.
Reaching these scales demands a fundamentally new approach to molecular synthesis. Implementing these approaches requires complex, tightly integrated hardware, often custom-developed for a specific application to address the requirements and challenges that arise. As AI models continue to expand the size of the design space that can be explored, this gap between computational capability and physical build capacity is likely to widen, increasing the strategic importance of scalable molecular synthesis platforms.
Reaction scaling is non-trivial and balances throughput, reagent costs, space requirements, technology platform accessibility, set-up and platform development investment cost. From the reagent consumption standpoint, current well-plate and column-based DNA and peptide synthesisers operate in reaction volumes of 10s-100s of μL, which translates to hundreds of litres — thousands of cubic meters of costly reagents, which would be required to synthesise DNA/peptides variant libraries to meet the needs of high-throughput screening/DNA data storage markets. In the current synthesis format, 10s-100s of μL of liquid are contained in individual DNA synthesiser columns or SBS1-sized multi-well plates, which occupy more space as the number of column/plate units increases, making the process extremely difficult to scale. The aforementioned constraints and high market demand for scalable DNA/RNA/peptide synthesis solutions call for alternative liquid-handling approaches. Ultimately, organisations without synthesis platforms that match the required scale, speed, and cost do not simply operate less efficiently. They operate within a narrower set of scientific and commercial strategies. That gap becomes harder to close as competitors who have solved the synthesis problem iterate faster, achieve greater diversity in sequence variants, and test across a broader range of what can be economically tested.
Liquid handling at ultralow volumes
Some attractive approaches to scale-out in biomolecular synthesis involve site-selective reagent activation with light, bead-based synthesis or synthesis in miniaturised emulsion droplets. These paths are scientifically elegant and, for certain applications, may eventually prove transformative. But each depends on deep innovation in chemistry, materials, optics and synthesis platform and are not yet mature enough to support commercial throughput at the cost points the market requires.
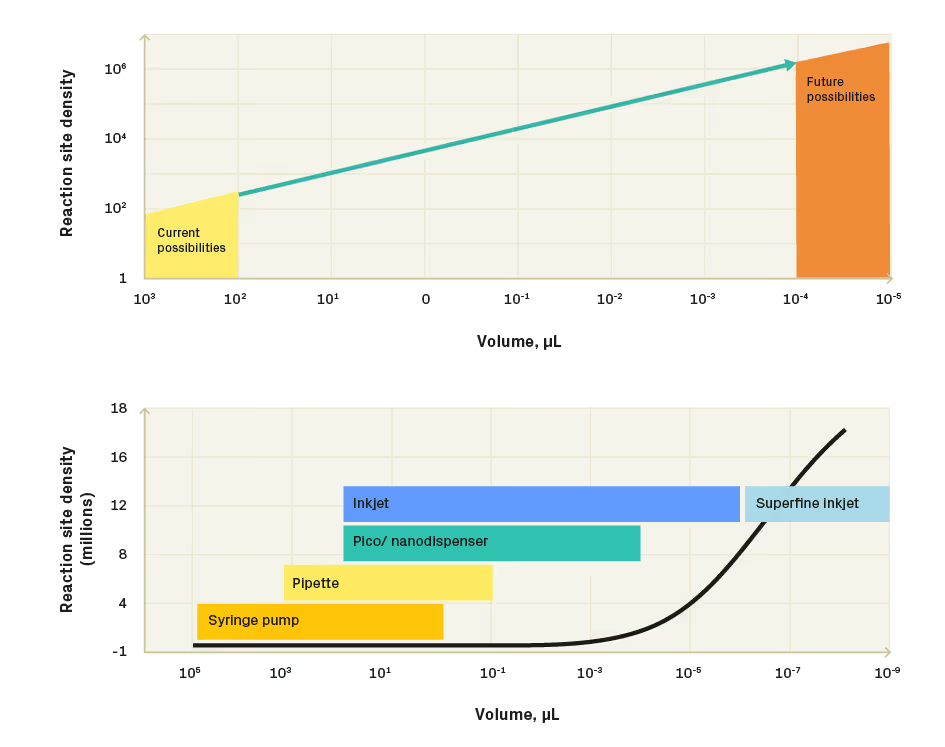
In contrast, a more straightforward route to managing space requirements and increasing reagent cost when scaling is to reduce reaction volume. For example, a 1,000,000-fold reaction volume reduction from 100 μL to 100 pL allows for an increase in reaction site density by 6000-fold (Figure 1 left). However, existing conventional liquid handling tools – syringe pumps, pipettes and pico-, nano dispensers - are no longer capable of transferring liquids in volumes lower than mid-pL range, way below the regime where significant scalability can be achieved (Figure 1 right). Accessing the regime with minimised reaction volumes and maximised reaction site density requires the implementation of orthogonal liquid dispensing approaches. Inkjet printing technology, which has been widely used in home/office printing and the graphics industry, has recently emerged as an attractive method for dispensing biological materials 2(cells, enzymes, nucleic acids, biological reaction buffers). Inkjet printers use rapid heating (thermal inkjet) or piezo actuators (piezo inkjet) to push tiny, low pL volume liquid drops through the nozzle in a highly controlled manner and deposit them on a substrate with precision as high as 21 μm. Due to the continuous nature of liquid dispensing, inkjet-dispensed volumes can be easily scaled up to hundreds of μL, enabling a very broad range of volumes that a single printing head can dispense, making inkjet a good choice for applications requiring a broad volume range and precise volume control. If an application requires sub-pL volume dispensing, superfine inkjet technology comes to mind. Superfine inkjet print heads can dispense inks down to fL volumes, pushing the boundary of what is possible even lower [3]. While inkjet offers a compelling route to low-volume dispensing, its successful application to biological systems depends strongly on managing fluid properties, nozzle reliability, and reagent stability during dispensing. Unlike industrial printing applications, biological reagents impose formulation constraints, shear-sensitivity requirements, and temperature tolerances that demand deliberate co-design of reagent and hardware.
Ultralow volume reaction format
In conventional applications, high-throughput screening and biomolecular synthesis are carried out in tubes, well plates, or microfluidic droplets. Automation solutions for handling tube racks and well plates include liquid handling robots. Some are very versatile, such as RAC modules from Ginkgo, Hamilton Star, Opentrons, Beckman, and Analytik Jena, and can be easily customised for specific assays or processes. Others are highly specialised for specific applications with minimised user hands-on time. Well-plate liquid-handling robots benefit from standardisation and versatility – they are compatible with standard labware and programmable directly by the end user. Another benefit of liquid handlers comes from liquid handler operational similarity to a manual benchtop process – both cases operate in a similar volume range, use a pipette to transfer liquids, and often the same type of reaction containers (multiwell plates, tubes), which makes the assay protocol transfer from benchtop to a robot much more straightforward. However, well-plate/tube liquid handlers do not scale – they quickly reach a limit on how many plates a single robot can handle at a time. Further scaling requires using multiple robots, which in turn limits how many instruments fit into a room, facility, or building (Figure 2).
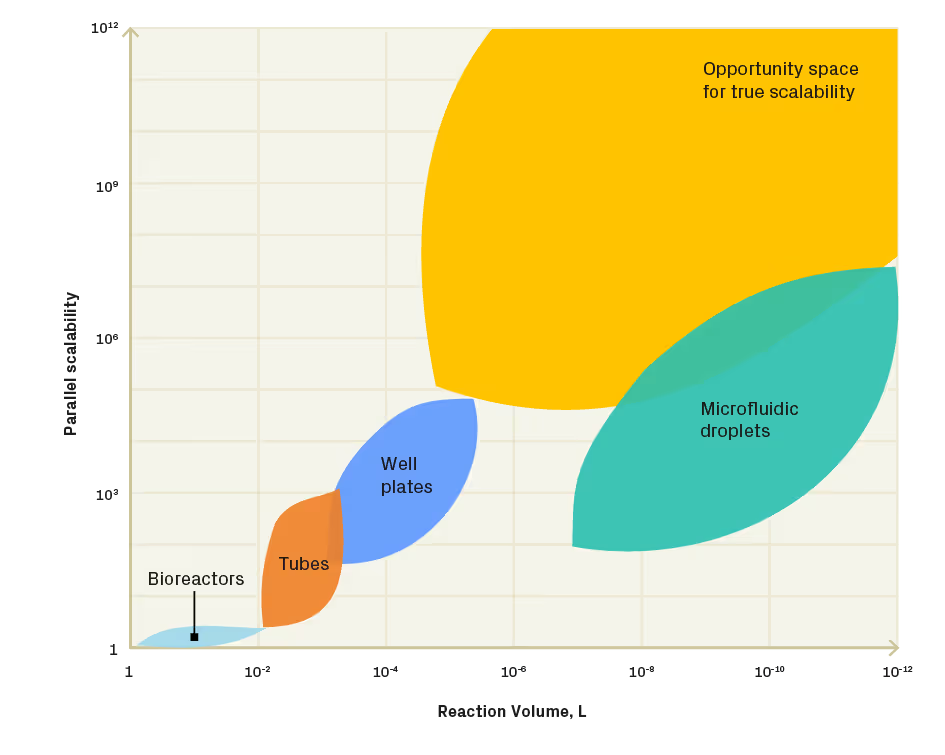
Inherent space constraints associated with well plates and conventional liquid handlers drive the need for alternative reaction formats. Alternative low-volume reaction formats include water emulsions in oil, generated using microfluidic droplet generation technologies. Flow-focusing and T-junction microfluidic devices can produce millions of pL-nL-volume drops per day. Each drop is a unique reaction vessel with enzymes, nucleic acids, reaction buffers or whole cells. Microfluidic drops can be manipulated by moving them in bulk fluid using conventional syringe pumps or individually on electrowetting chips, enabling operations such as sorting, merging, splitting, mixing, and incubation [4]. Drop compatibility with conventional light detection techniques, such as absorbance or fluorescence, makes this reaction format very attractive for high-throughput screening applications. Microfluidic drops are compact and can be stored and manipulated in small containers – an emulsion of a few millions of pl drops fits into a single 1.5 mL Eppendorf tube, making this reaction format easily scalable for parallel processes (Figure 2). Microfluidic drops are well-suited for applications requiring high-throughput processing and scalability. However, because water-in-oil drop emulsions are moved by bulk fluid flow, it becomes extremely challenging to re-address individual drops. This limitation may be critical in applications that require addressing each reaction site multiple times, such as DNA oligo synthesis.
Perhaps the simplest reaction format that overcomes some limitations of microfluidic drops is small reaction drops on a flat substrate. When combined with precise ultralow volume liquid dispensing technologies, such as inkjet, flat substrates offer a range of advantages:
- Simple to manufacture and handle
- Compatible with ultralow volume dispensing technologies
- In situ optical reaction QC is possible due to substrate transparency
- Reaction sites are physically separated – each site can be addressed multiple times
- Each site can be easily molecularly barcoded for downstream applications
- High reaction site density – millions of reaction sites can be packed in a small area
- Ultimate scalability – flat substrates can take up a variety of shapes and forms, from small microscopy glass slides to large glass, plastic sheets or flexible tape rolls, offering different scalable substrate packing options
- Wider range of opportunities for substrate washing between reaction cycles – unlike well plates, flat substrates do not rely on pipettes for reaction site washing. Bath, continuous liquid flow, or other novel low-fluid-consumption washing approaches could be used for this purpose